|
Lowcost-Rektifikation mit der Chembox: Der Topfreiniger als Füllkörper Viktor Obendrauf Download des Artikels als geziptes Winwordfile (73 kByte)
Unabhängig von den historischen Dimensionen zählen Destillationen und Rektifikationen heute zu den bedeutendsten Methoden zur Trennung und Reinigung von flüssigen Stoffsystemen in Labor und Technik. Die destillative Reinigung im präparativen Labor, die Fraktionierung von Erdöl in der Raffinerie und die Erzeugung von hochprozentigen alkoholischen Getränken sind nur einige Beispiele, die auch in der Schule immer wieder angeführt werden. Nach einer möglichst knapp gehaltenen Zusammenfassung der physikalischen Grundlagen werden im experimentellen Teil Versuchsanleitungen zum Thema Destillation und Rektifikation mit einfachsten Mitteln vorgestellt. Mit Hilfe des komfortablen Meßwerterfassungssystems Chembox/Chemex (IBK) wird gezeigt, daß die Trennwirkung einer Halbmikro-Destillationsapparatur (Schliffapparatur ohne Kolonne aus der Schulausstattung) vergleichsweise schlechter ist als die zur Diskussion gestellte Low-Cost-Version mit improvisierter "Kolonnenfüllung". Einige Vokabel... Bei einer einfachen Gleichstrom-Destillation wird in einem Verdampfer-Kolben (Blase) eine Flüssigkeit zum Sieden gebracht und der Dampf in einem Kühler (Kondensator) wieder als Destillat kondensiert. Bei einer fraktionierten Destillation wird das Destillat nach steigendem Siedepunkten in mehreren Fraktionen gesammelt. Bei einer Gegenstrom-Destillation oder Rektifikation wird ein Teil des kondensierten Dampfes in der Destillationskolonne, die sich zwischen Verdampfer und Kondensator befindet, als Rücklauf dem aufsteigenden Dampf entgegengeführt. Dabei findet ein Stoffaustausch zwischen beiden Phasen statt: Die leichter flüchtigen Komponenten reichern sich in Richtung des Kondensators im Dampf an, die schwerer flüchtigen Komponenten konzentrieren sich im Rücklauf. Die Kolonnendestillation stellt in gewisser Weise eine Kaskade von unmittelbar hintereinander wiederholten Gleichstromdestillationen dar, wobei der Trenneffekt einer einzigen, einfachen Gleichstromdestillation als sogenannter "theoretischer Boden" bzw. als "Trennstufe" bezeichnet wird. Die Effektivität einer Destillation hängt von vielen Faktoren ab:
und Stoffdurchsatz. Die physikalisch-chemischen Grundlagen Bei Destillationen und Rektifikationen nützt man die unterschiedlichen Flüchtigkeiten bzw. Siedetemperaturen der im Stoffgemisch enthaltenen Reinstoffe. Als Maß für die Flüchtigkeit eines Reinstoffes kann der jeweilige Dampfdruck über diesem Reinstoff angesehen werden. Der Dampfdruck eines Stoffes steigt mit der Temperatur stark an. So hat Eis bei 0°C einen Dampfdruck von etwa 6 mbar, Wasser von 20°C hat einen Dampfdruck von ca. 23 mbar, 80°C heißes Wasser hat bereits einen Dampfdruck von etwa 474 mbar. Bei 100 °C beträgt der Dampfdruck von Wasser 1013 mbar. Die Siedetemperatur einer reinen Flüssigkeit ist bekanntlich dann erreicht, wenn der Dampfdruck des Reinstoffs dem Umgebungsdruck entspricht. Die Temperaturabhängigkeit des Dampfdrucks (und damit auch des Siedepunkts) eines Reinstoffes wird durch die CLAUSIUS-CLAPEYRONsche Gleichung beschrieben. Quantitative Aussagen dazu findet man in der einschlägigen Literatur [1,2]. Eine genauere mathematische Beschreibung würde den Rahmen dieser Vorbemerkungen zu den folgenden Low-Cost-Versuchsbeschreibungen sprengen Grob kann gesagt aber werden, daß eine Halbierung des äußeren Drucks die Siedetemperatur eines Reinstoffes um etwa 15°C senkt. Thermisch instabile Stoffe können bekanntlich im Vakuum schonender destilliert werden. Ein einfacher Low-Cost-Versuch zur Demonstration der Siedtemperaturerniedrigung bei Druckerniedrigung findet sich im experimentellen Teil dieses Beitrags. Die Trennbarkeit eines Stoffgemisches wird bestimmt vom Dampf-Flüssigkeits-Gleichgewichtsverhalten. Für sogenannte ideale Gemische läßt sich der Zusammenhang zwischen den zusammengehörigen Konzentrationen der Flüssigkeiten und Dampfphase aus den Dampfdrücken bzw. Flüchtigkeiten der reinen Komponenten berechnen. Den Zusammenhang zwischen dem Partial-Dampfdruck einer Komponente im Dampfraum und der Konzentration der Komponente in der flüssigen Mischung in Abhängigkeit vom Dampfdruck der reinen Komponente skizziert im Idealfall eines binären Gemisches das RAOULTsche Gesetz:
pA und pB = Partialdrücke der beiden Komponenten A und B im Dampfraum xA und xB = Stoffmengenanteile der Komponenten A und B in der Flüssigkeit PA und PB = Dampfdrücke der reinen Komponenten A und B Stoffmengenanteil: Konzentrationsangabe in Mischphasen; definiert als Quotient aus Zahl der Mole (Stoffmengenanteil) einer Komponente und Gesamtzahl der Mole in der Mischung. Wenn z. B. nur eine Komponente vorliegt, beträgt für diesen Stoff der Stoffmengenanteil genau 1, für die zweite Komponente 0. Aus den Gesetzen (1) und (2) läßt sich durch Umformung und Einsetzen nach [1] folgende Beziehung ableiten:
Der Quotient der Dampfdrücke der reinen Komponenten wird auch als relative Flüchtigkeit a
bezeichnet:
y = Stoffmengenanteil der leichter flüchtigen Komponente A im Dampfraum x = Stoffmengenanteil der leichter flüchtigen Komponente A in der Flüssigkeit PA = Dampfdruck der reinen (leichter flüchtigen) Komponente A PB = Dampfdruck der reinen Komponente B Gemäß obiger Gleichung müssen PA und PB unterschiedlich sein, damit sich Dampfphase und Flüssigkeit in ihrer Zusammensetzung unterscheiden und eine Destillation überhaupt möglich ist. Die Anreicherung der leichter flüchtigen Komponente im Dampfraum ist umso größer, je unterschiedlicher die Dampfdrücke der reinen Komponenten sind. Unterscheiden sich die Dampfdrücke der zu trennenden Stoffe nur wenig, so kann man mit einer einmaligen Verdampfung (und anschließender Kondensation) keine ausreichende Trennung erzielen. In diesen Fällen wird der Verdampfungsprozeß mehrmals wiederholt, was in Destillationskolonnen automatisch geschieht. Als Grundregel nach [1] gilt, daß bei einer Siedepunktsdifferenz von weniger als 80°C ohne Destillationskolonne keine effiziente Trennung mehr möglich ist. Der azeotrope Alkohol (Siedetemperatur ca. 78°C) läßt sich somit ohne Fraktionierkolonne nicht mehr effizient aus einer wässrigen Lösung abtrennen. Die Aufnahme einer Siedekurve (Siedetemperatur über Destillatmenge) erscheint in jedem Fall nützlich. Das RAOULTsche Gesetz beschreibt die Anreicherung der leichter flüchtigen Komponente im Dampfraum durch eine einmalige Verdampfung (einfache Gleichstromdestillation). Nach n-maliger Wiederholung des Verdampfungs-Kondensations-Vorganges und Einsetzen der Gleichung (4) hält man nach [1]:
yn = Stoffmengenanteil der leichterflüchtigen Komponente im Dampfraum nach n-maliger Verdampfung x1 = Stoffmengenanteil der leichter flüchtigen Komponente in der flüssigen Phase n = Zahl der Wiederholungen des Verdamfpungsvorganges (Zahl der theoretischen Böden für die Trennung eines binären Gemisches). Aus Gleichung (5) ist ersichtlich, daß durch mehrmalige Wiederholung des Verdampfungs- und Kondensationsvorganges eine Potenzierung der Trennwirkung erzielt werden kann. Aus Gleichung (5) läßt sich nach Umformung die Zahl der notwendigen theoretischen Böden für eine vorgegebene binäre Mischung und gewünschte Destillatzusammensetzung berechnen. Auch eine graphische Bestimmung ist möglich: 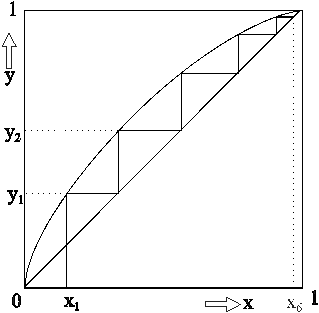 Auf der y-Achse sind die Stoffmengenanteile y der leichter flüchtigen Komponente in der Dampfphase von 0 bis 1 aufgetragen . Die x-Achse zeigt die Stoffmengenanteile x der leichter flüchtigen Komponente in der flüssigen Phase ebenfalls von 0 bis 1. Wenn die relative Flüchtigkeit mit a = 1 angenommen wird (keine Dampfdruckunterschiede PA und PB zwischen den Komponenten), dann entspricht die Gleichung (5) der Geraden y = x durch den Ursprung mit einer Steigung von 45° im Diagramm. Je größer nun die relative Flüchtigkeit a ist, desto stärker nach oben gekrümmt ist die Gleichgewichtskurve. Wenn nun z. B. ein flüssiges Gemisch mit der Zusammensetzung x1 (Stoffmengenanteil der flüchtigeren Komponente in der Flüssigkeit) verdampft wird, befindet sich in der Dampfphase der flüchtigere Stoff mit der größeren Konzentration y1. Diese Konzentration y1 bleibt natürlich bei der Kondensation erhalten (y1 = x2). Nach neuerlicher Verdampfung wird in der Dampfphase bereits die Konzentration y2 erreicht usw. Nach insgesamt fünfmaliger Wiederholung (= 5 theoretische Böden) ist die flüchtigere Komponente in der Flüssigkeit von der Konzentration x1 auf die Konzentration x5 angereichert worden (siehe Diagramm). Da sich bei den dargestellten Verhältnissen um Gleichgewichtskonzentrationen handelt, gilt das Diagramm nur dann, wenn bei der Rektifikation kein Destillat abgenommen wird. Das gesamte Kondensat ist einem Rückfluß unterworfen. In der Praxis wird dieses Gleichgewicht jedoch ständig gestört. Im schlechtesten Fall ist der Rücklauf (z. B. durch Überhitzung der Kolonne) überhaupt unterbrochen, die Kolonne "staut". In diesem Fall ist eine reguläre Rektifikation nicht mehr möglich. In der Praxis muß somit ein optimierter Mittelweg beschritten werden: Durch Einführung des sogenannten "Rücklaufverhältnisses" (= Quotient aus Rücklauf und Destillat) und einigen Umformungen gelangt man nach [1] schließlich im dargestellten Diagramm zu einer neuen Geraden ("Arbeitslinie"), die nun die Gerade y = x ersetzt. Das Rücklaufverhältnis muß nun mindestens so groß gewählt werden, daß die resultierende neue Gerade in der Graphik eine gewünschte Trennung überhaupt noch theoretisch möglich macht. Auf diese Weise läßt sich das "Mindestrücklaufverhältnis" ermitteln. Im Idealfall (totaler Rücklauf) geht die "Arbeitslinie" in die theoretische Gerade y = x über. Unter diesen Bedingungen kann die "Mindestbodenanzahl" für eine gewünschte Trennung eruiert werden. Je größer das Rücklaufverhältnis bei einer Rektifikation, desto weniger theoretische Trennstufen sind notwendig und umgekehrt. Bei vielen Stoffsystemen treten zusätzlich beträchtliche Abweichungen vom idealen Verhalten der Gleichgewichtskonzentrationen auf, sodaß auch der Verlauf der gekrümmten Kurve durchaus nicht symmetrisch ist. Beim binären System Ethanol/Wasser z. B. schneidet die gekrümmte Kurve die Gerade y = x bei einer Konzentration von 96% Alkohol. Eine Anreicherung der Dampfphase mit der flüchtigeren Komponente für eine destillative Trennung ist somit bei dieser Zusammensetzung des Alkohol/Wasser-Gemisches nicht mehr möglich. Bekanntlich bildet Alkohol dieser Konzentration mit Wasser ein sogenanntes Azeotrop. Die technischen Lösungen Bei industriellen Anlagen, die naturgemäß auf niedrige Betriebskosten ausgelegt sind, strebt man ein niedriges Rücklaufverhältnis mit vergleichsweise hohen theoretischen Trennstufen an. Die apparativen Voraussetzungen dafür liefern zum Beispiel Bodenkolonnen und Oberflächenrektifikatoren (Füllkörpersäulen, Rieselsäulen). Bei den Bodenkolonnen (Glockenböden, Siebplatten etc.) wird die im jeweiligen Boden angesammelte flüssige Phase von Dämpfen aus dem nächsttieferen Boden durchströmt. Flüchtige Anteile gelangen in die Gasphase und weiter in den nächsthöheren Boden. Bodenkolonnen werden heute mit Durchmessern von wenigen Zentimetern bis zu 12 Metern (!) mit praktischen (tatsächlichen) Bodenzahlen über 200 gebaut. Bei den Oberflächenrektifikatoren handelt es sich um Benetzungssäulen mit zwei geschlossenen Phasen, die sich gegeneinander bewegen, ohne sich zu durchdringen. Zwischen den beiden gegenläufigen Phasen kommt es zum Stoffaustausch zwischen Dampf und Flüssigkeit durch Diffusion an der Phasengrenze. Für eine entsprechend große Phasengrenzfläche sorgen bei Füllkörpersäulen sogenannte Schüttfüllungen (Füllkörper aus Keramik, Metall, Glas etc), über die sich ein Flüssigkeitsfilm ausbreiten kann. Je kleiner und gleichmäßiger die Füllkörper sind, desto besser ist die Trennleistung, weil Stoff- und Wärmeaustauschwirkung auf eine größere Oberfläche verteilt wird und der durchströmende Dampf immer wieder abgelenkt und neu verteilt wird. Die Einsatzbereiche von Glockenboden- und Füllkörperkolonnen haben dort ihre Grenzen, wo man aus thermischen Gründen mit niedrigen Dampfdrücken arbeiten muß. In solchen Fällen arbeitet man mit Rieselkolonnen (= nicht gefüllte Kolonnenrohre mit geringem Durchmesser), in denen die Dämpfe ohne wesentlichen Druckverlust nach oben strömen, während der Rücklauf an der Kolonnenwand einen nach unten wandernden Film bildet. Destillieren mit der CHEMBOX Selbstverständlich soll die klassische Destillationsapparatur mit Claisenaufsatz bei Verfügbarkeit an geeigneter Stelle in den jeweiligen Schulstufen auch eingesetzt werden. Die Trennleistung derartiger Destillationsbrücken nach Claisen, die an Österreichs (Höheren) Schulen meist aus Halbmikro-Schliffgerätesätzen zusammengebaut werden, darf jedoch nicht überschätzt werden. Ohne zusätzliche Fraktionierkolonne (in den Bausätzen meist vorhanden) und entsprechend sorgfältiger und damit aber zeitraubender Temperaturführung ist zum Beispiel eine Abtrennung des azeotrop bei 78°C siedenden Alkohols aus einer wässrigen Lösung nicht machbar. Diese Tatsache wird deutlich sichtbar, wenn während der Destillation ein klassisches Siedediagramm (Siedetemperatur/Destillatmenge-Diagramm) angefertigt wird. Durch die erfreulich komfortablen Möglichkeiten der automatischen Meßwerterfassung mittels Computer und angeschlossener CHEMBOX (IBK) läßt sich unter Verzicht auf den Tropfenzähler zur Volumsmessung analog dem klassischen Siedediagramm praktisch "nebenbei" ein ebenfalls recht aufschlußreiches Siedetemperatur/Zeit-Diagramm erstellen, wenn man auf der Abszisse das Destillatvolumen durch eine einfache Zeitachse ersetzt. Das folgende Diagramm entstand bei der Destillation von 50 ml Rum (38 Vol%) mit einer einfachen Halbmikro-Destillationsapparatur. Zwecks Verkürzung der gesamten Versuchsdauer entsprechend den Erfordernissen des Schulalltags wurde kein elektrischer Heizpilz als Energiequelle verwendet, sondern ein Laborbrenner mit Drahtnetz (Heißluftbad). Geräte: 100 ml Destillationsrundkolben NS 19 (inkl. Klemme) Claisenaufsatz NS 19 mit Stopfen und Schraubverschluß mit verschiebbarem Thermometer Liebig-Kühler NS 19 inkl. Schlauchverbindungen und Klemme Destilliervorstoß NS 19 inkl. Klemme 50 ml Rundkolben NS 19 inkl. Klemme Thermometer (bis 100°C) Siedesteinchen Stativschiene, 2 Stativstangen, 2 Muffen, 1 Klemme, 1 Eisenring, Brenner Zur elektronischen Temperaturmessung: Notebook IBM Thinkpad 340 CSE (486er) Chembox mit Software Chemex (IBK) Portabler Drucker Canon BJ 30 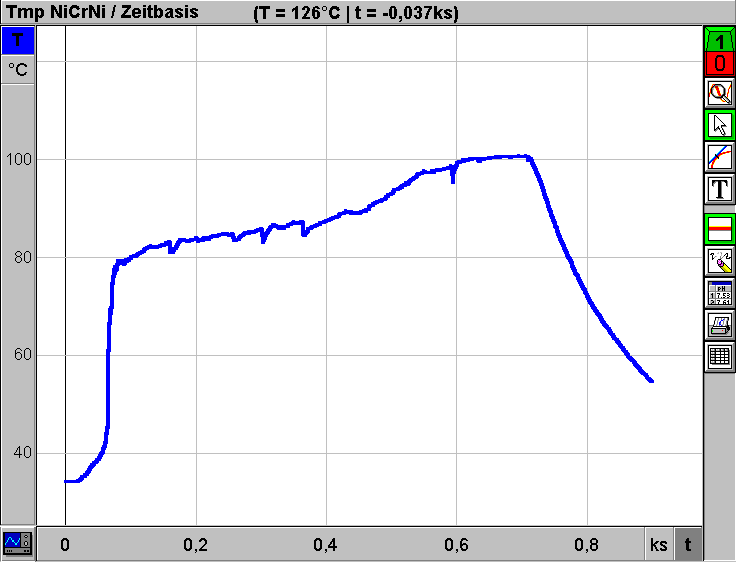 Die Kosten für die beschriebene (variable) Destillationsapparatur (Glasgeräte) betragen mehr als 1.600.-inkl. MWSt. Das verwendete Stativsystem inkl. Schienenstativ, Stativstangen, Klemme, Ring und Muffe kommt bei Einzelbestellung aus einem Lehrmittelkatalog auf etwa 1500.- inkl. MWSt. Das Notebook (mit großer Chemex-Meßwertanzeige am Notebook-LCD) entspricht mit der problemlos anschließbaren Chembox vom Handling fast einem konventionellen Multimeter mit LED. Mit dem Ausdruck der Schreiberdarstellung mittels transportablem Drucker wird jedoch das Temperatur/Zeit-Diagramm zusätzlich allen Schülern der Klasse sofort zugänglich. Wie man dem Schreiber-Diagramm entnehmen kann, wurde für eine zügige Erwärmung der Rumprobe gesorgt. Nach etwa 1,5 Minuten (0,1 ks) erreicht der Alkoholdampf das Thermometer bzw. den Sensor. Für das Abdestillieren der im vorgelegten Rum (50 ml) enthaltenen Menge Ethanol (ca. 20 ml) wurde dem System nochmals über 8 Minuten (bis 0,6 ks) Zeit gelassen. Wie der mehr oder weniger stetige Anstieg der Dampftemperatur von etwa 80°C bis 100°C beweist, ist die Trennwirkung unter diesen Bedingungen nicht sehr befriedigend.
Destillieren aus dem Reagenzglas Die einfache Überprüfung von Trennleistungen diverser Destillationstechniken mit Hilfe der elektronischen Meßwerterfassung kann auch auf Low-Cost-Destillationsgeräte übertragen werden. In Weiterführung der Anregungen zum Experimentieren mit einfachen Geräten zur Einsparung von Zeit und Material-Ressourcen [3,4,5,6,] wird im folgenden eine "Apparatur" zur Diskussion gestellt, bei der nur zwei Reagenzgläser als Glasgeräte verwendet werden: Geräte: 2 Reagenzgläser (Fiolax 16/160) ca. 7 cm langes Stück Topfreiniger (Stahldrahtlappen) als "Kolonnenfüllung" 2 Reagenzglashalter 4 Kanülen (0,8/120 mm) als Kühler 1 Weichgummistopfen (grau, 18D) Siedesteinchen Bunsenbrenner Geräte zur elektronischen Meßwerterfassung: siehe oben 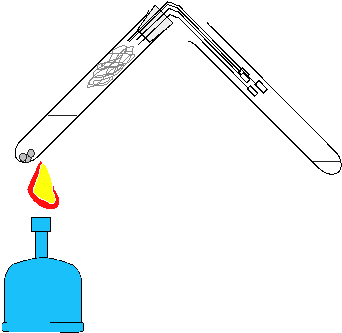 Durchführung: Ein Reagenzglas wird ca. 5-6 cm hoch mit Rum (38%ig) und einigen Siedesteinchen beschickt. Nun steckt man ein ca. 7cm langes Stück eines Metall-Topfreinigers ("Putzwaschel") dicht gepackt in das Reagenzglas. Das Reagenzglas wird mit dem Gummistopfen verschlossen, in dem sich die vier Kanülen als Kühler befinden. Die Kanülen müssen in unterschiedlicher Länge durch den Stopfen gestochen und rechtwinkelig gebogen werden, damit alle vier Luer-Ansätze wie in der Skizze gezeigt möglichst tief ins zweite Reagenzglas hineingesteckt werden können. Mit zwei Reagenzglashaltern kann die Apparatur in einer Hand gehalten werden. Nun wird vorsichtig mittels Kartuschenbrenner für eine Erwärmung gesorgt. Sobald die Flüssigkeit zu sieden beginnt, muß die Wärmezufuhr gedrosselt werden, damit die "Kolonne" nicht zu stauen beginnt. Zur Messung der "Kolonnenkopf"-Temperatur wird der dünne NiCrNi-Drahtsensor seitlich am Stopfen vorbei im Reagenzglas positioniert. Wie das mit dieser Apparatur (Gesamtkosten inkl. Reagenzglashalter ca. 40.-) aufgenommene Diagramm zeigt, wird selbst bei verkürzter Versuchszeit eine effiziente Trennung erzielt: 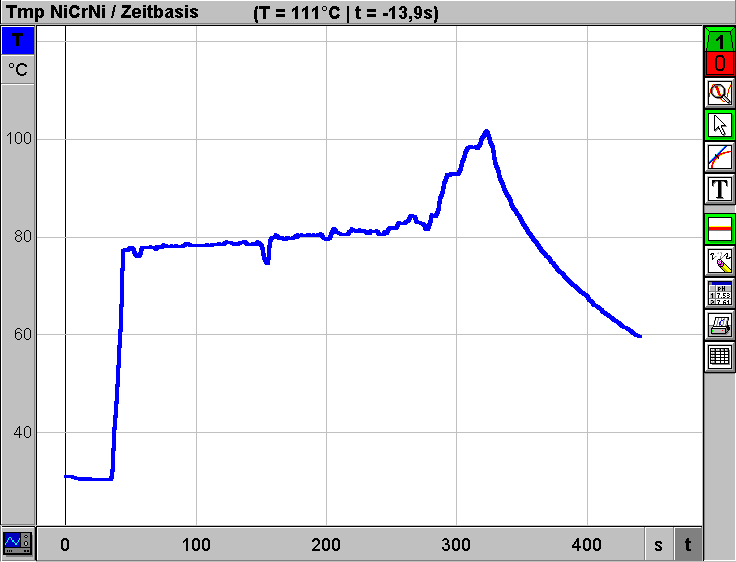 Die Effizienz der "Kolonnenfüllung" wird im Vergleich erkennbar. Das folgende Diagramm wurde unter den gleichen Bedingungen mit der gleichen Low-Cost-Apparatur, jedoch ohne "Putzwaschel"-Füllung erhalten: 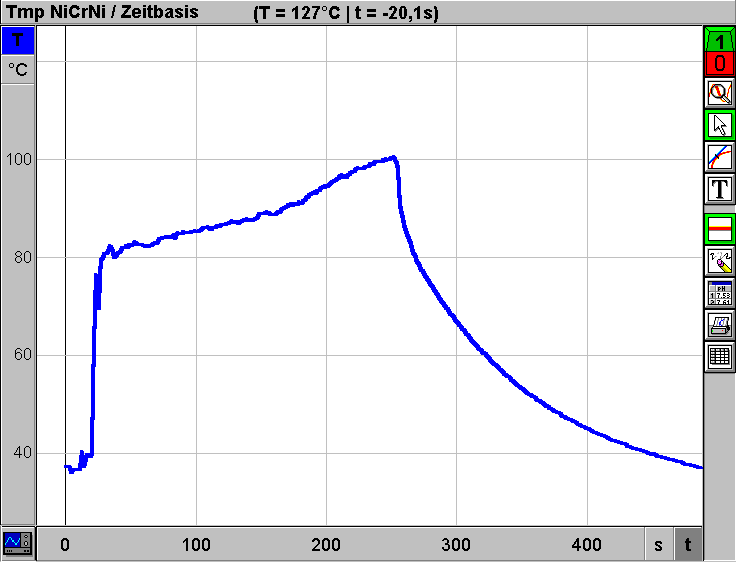 Der prinzipielle Unterschied zur Siedekurve, die mit der konventionellen Halbmikroapparatur erhalten wurde, liegt eigentlich nur im unterschiedlichen Zeitbedarf: Die geringe Menge an Alkohol im Reagenzglas kann ohne "Putzwaschel"-Kolonne in ca. 4 Minuten (250 s) abdestilliert werden. Hinweise zum "Kanülenkühler": Die Kanülenspitzen werden nach dem Durchstechen des Stopfens sofort entschärft, indem man die abgeschrägten Enden mittels Seitenschneider kappt. Die Kanülen halten unter den beschriebenen Versuchsbedingungen jahrelang, wenn man sie nach Ende des Versuchs mittels durchgesaugter Luft auch innen trocknet. Dichteprüfung des Destillats aus der Low-Cost-Destillation Da die beschriebenen Destillationen aus dem Reagenzglas sehr rasch durchführbar sind, kann die gewonnene Zeit für weiterführende Untersuchungen sogar im Rahmen einer Schulstunde genützt werden: Die Effizienz des "Putzwaschels" als Low-Cost-Kolonne läßt sich mittels Dichteprüfung des Destillats abschätzen, wenn nur jenes Destillat gesammelt wird, das um etwa 80°C kondensiert werden kann: Geräte (siehe auch Titelfoto dieser Zeitschrift): Waage (VCÖ-Aktionswaage Kern, 0,01g Ablesegenauigkeit siehe Inserat und Titelseite) 2-ml-Spritze (HSW Normject) 1 Kanüle (0,8/120mm) Reagenzglas mit Destillat aus Low-Cost-Destillation Durchführung: Die 2-ml-Spritze wird ohne Kanüle auf die Waage gelegt, nach Tarierung saugt man mittels Kanüle langer Kanüle genau 1 cm3 Destillat aus dem Reagenzglas, entfernt die Kanüle und bestimmt die Masse von 1 cm3 Destillat. Die erhaltenen Meßwerte liegen um 0,80g/cm3 (Zum Vergleich: Dichte von 80%igem Ethanol bei 20°C 0,84494 g/cm3, Dichte von 90%igem Ethanol bei 20°C 0,81942 g/cm3, Dichte von 96%igem Alkohol: 0,80280 g/cm3 ) Low-Cost-Siedepunkterniedrigung Wenn im Zuge der Besprechung von Destillationen die Siedepunktserniedrigung für thermisch instabile Systeme durch Druckverminderung rasch und doch überzeugend gezeigt werden soll, kann man folgendermaßen vorgehen: Geräte: 1 Reagenzglas (Fiolax 16/160) 1 Weichgummistopfen (grau, 18D) mit 1 Kanüle 1,2/40 mm (Spitze entschärft) 1 20-ml-Spritze (HSW-Softjekt mit Gummidichtung) Siedesteinchen Wasser Brenner Durchführung: Das Reagenzglas wird ca. 5 cm hoch mit normalem Wasser aus der Wasserleitung beschickt. Nach Zugabe von einigen Siedesteinchen (!) erhitzt man mittels Brenner bis zum Sieden. Nach Entfernung des Brenners wird das Reagenzglas rasch mit dem Stopfen inkl. Kanüle verschlossen. Wenn man nun mittels aufgesetzter Spritze einen entsprechenden Unterdruck erzeugt, beginnt das fast noch 100°C heiße Wasser im Reagenzglas wieder für kurze Zeit heftig zu sieden. Wenn man den gesammelten Dampf in der Spritze rasch entleert und mit der Spritze wieder einen Unterdruck erzeugt, kann das Sieden nochmals beobachtet werden. Hinweis: Die Demonstration von "Kelomat"-Verhältnissen (höhere Siedetemperatur bei höherem Druck) sollte mit der beschriebenen Apparatur durch Festhalten des Spritzenstempels aus Sicherheitsgründen nicht gezeigt werden. Die Gefahr, daß dabei der Stopfen nicht im Reagenzglas bleibt, ist zu groß. Unkontrolliertes Austreten von kochendem Wasser bzw. von überhitzten Wasserdampf wäre die Folge.
Viktor Obendrauf e-mail: viktor@obendrauf.com
|
| update: 02.02.2021 zurück zur Hauptseite |
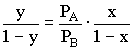 (3)
(3)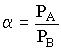 (4)
(4)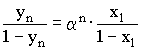 (5) = relative Flüchtigkeit
(5) = relative Flüchtigkeit